







 Tandy Warnow, David Bruton, Jr. Centennial Professor in Computer Science.Scientists continue to refine, and sometimes radically alter, our understanding of the “Tree of Life” — the ways in which species are related to one another. They’re using the computing power of the Texas Advanced Computing Center (TACC) at The University of Texas at Austin to better understand the origin of species and, ultimately, help fight disease and develop better crops.
Tandy Warnow, David Bruton, Jr. Centennial Professor in Computer Science.Scientists continue to refine, and sometimes radically alter, our understanding of the “Tree of Life” — the ways in which species are related to one another. They’re using the computing power of the Texas Advanced Computing Center (TACC) at The University of Texas at Austin to better understand the origin of species and, ultimately, help fight disease and develop better crops.
Whereas once, evolutionary history was based on the relationships of bones, skeletons and other morphological clues, today DNA is now the main informer in the story of how the Earth became such a diverse place.
Phylogenetics is the branch of life science that studies the evolutionary relationships among organisms based on genetic evidence. By aligning the molecular sequences of different species, scientists can see how organisms differ at the genetic level, determine where they diverged and map out branching trees of relationships based on the alignments.
With the cost of gene sequencing declining, researchers are performing more phylogenetic studies. Even so, the process of lining up tens of thousands of sequences from hundreds or thousands of species is incredibly complicated, even for a computer.
“The most accurate trees are estimated using methods that try to solve hard optimization problems,” said Tandy Warnow, professor of computer science at The University of Texas at Austin and a Guggenheim Fellow.
“While those solutions can be done on small data sets or moderate sized data sets, on large data sets, they can take a very long time — weeks to months to years of computational time. The Texas Advanced Computing Center ends up being essential for those problems.”
TACC, on the J.J. Pickle Research Campus in north Austin, runs some of the biggest and most powerful systems in the world, but even their supercomputers can hardly keep up with the pace of genetic research. According to Moore’s law, the performance of computers doubles every two years. However, the ability of gene sequencers to create data has grown at an even faster rate.
“It’s a different kind of challenge,” Warnow said. “It’s not just how we run analyses on big data sets, but how do we access the data in a way that is sensible?”
Divide and Conquer
Warnow is working with postdoctoral fellow Kevin Liu of Rice University and Siavash Mirarab, a Ph.D. student in computer science at The University of Texas at Austin, to create smarter, faster and more accurate algorithms to apply to some of the biggest data sets ever created.
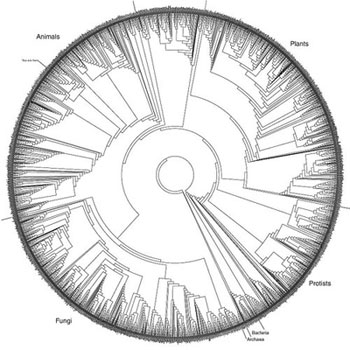 This phylogenetic tree, created by David Hillis, Derreck Zwickil and Robin Gutell, depicts the evolutionary relationships of about 3,000 species throughout the Tree of Life. Less than 1 percent of known species are depicted.With a $1.5 million grant from the National Science Foundation (through the Assembling the Tree of Life project), the researchers have developed software that allows computers to draw better evolutionary trees more quickly.
This phylogenetic tree, created by David Hillis, Derreck Zwickil and Robin Gutell, depicts the evolutionary relationships of about 3,000 species throughout the Tree of Life. Less than 1 percent of known species are depicted.With a $1.5 million grant from the National Science Foundation (through the Assembling the Tree of Life project), the researchers have developed software that allows computers to draw better evolutionary trees more quickly.
It’s called SATé — Simultaneous Alignment and Tree Estimation — and uses a novel divide-and-conquer approach.
“By dividing a really big data set that’s hard to align into small data sets that are closely related, you can get good estimates on each subset and then get an alignment on the full data set,” Warnow explained.
Massive supercomputers, such as Ranger at TACC, align the sequences of each subset and combine the alignments into an alignment on the full set of sequences.
There’s no way to know whether the tree that emerges from these simulations is absolutely accurate. Some trees are obviously wrong — for example, those that show humans and crocodiles on the same branch, separated from chimps — but most are probable.
For that reason, SATé uses a statistical method to provide a maximum likelihood score: a measure by which to assess its accuracy against other answers. SATé repeats the process of alignment and tree-building many times until a tree with the highest likelihood score is reached.
In software development, the best products are not just the newest, but the ones that are proved to be better than the alternatives. To this end, Warnow and her team have been working as quality assurance and reliability testers, solving hard evolutionary tree problems multiple times, with different methods and parameters, to ensure that SATé produces the highest-quality result.
First reported in the journal Science and later explored in the journals PLoS Currents and Systematic Biology, the researchers have shown repeatedly that SATé works as well as the alignment and tree estimation methods that are commonly used, which analyze trees as single units. But SATé is far faster or achieves greater accuracy but in the same amount of time.
For the Birds
Warnow and her team also collaborate with evolutionary biologists on projects in which their guidance can lead to new insights.
Since Charles Darwin’s day, scientists have debated the evolutionary history of flightless birds, known at ratites. How did so many similar species get to such far-flung corners of the Earth?
“The theory of continental drift provided a convenient answer,” said Michael Braun, a curator in the department of systematic biology at the Smithsonian Institution. “These birds evolved from a common flightless ancestor and then drifted to their current distributions. For 40 years, this remained the textbook explanation of species dispersal.”
That is until Braun discovered through DNA analysis that an ancient (but still living) family of birds found in South America, the tinamou, was one of the most closely related groups to emus and ostriches. But the tinamou could fly — a finding first reported in 2009.
This fact, combined with the lack of skeletal evidence of flightless birds before the continents broke apart, led to a re-conceptualization of the ratite branch of the avian tree. Ratites were in fact descended from flying birds that traveled to places where flight was no longer an evolutionary advantage and consequently lost their ability to fly.
“It‘s hard to recognize the relationships among species using just morphology, but when we can use the molecules and appropriate analytical methods to find the relationships, it helps us understand better how that adaptive evolution has occurred,” Braun said.
Recently, Warnow worked with Braun, using SATé, to reanalyze his controversial findings. Their study confirmed the evolutionary relationship that Braun found.
Emergency Phylogenetics
Better, faster, more accurate phylogenetic methods can have a life or death impact for humans.
The Centers for Disease Control and Prevention uses sequence alignment and evolutionary tree-building tools when a new virus emerges to determine where it might have come from and how it differs from previous viruses.
Plant scientists also use tree-building tools to determine which genes are associated with positive traits such as hardiness and drought tolerance. This knowledge is enabling scientists to breed more productive crops, helping to feed the world.
But none of these problems is easily solved.
“Many research groups are estimating trees containing anywhere from a few thousand to hundreds of thousands of species, towards the eventual goal of estimating a Tree of Life, containing perhaps as many as several million leaves,” Warnow wrote in a recent article in Systematic Biology. “These phylogenetic estimations present enormous computational challenges, and current computational methods are likely to fail to run even on datasets in the low end of this range.”
In other words, small problems may be within reach, but the big ones remain.
“It’s not getting any easier, but it is getting more fun,” Warnow said.
By Aaron Dubrow, originally published on the Texas Advanced Computing Center website.









Comments